Av Åsmund Flobak, lege i spesialisering i onkologi, St. Olavs hospital, Trondheim, postdoc NTNU

Åsmund Flobak
Hvorfor har høye mennesker større sjanse for å få kreft enn lave mennesker? Hvorfor har vi mer basalcellekarsinom enn malignt melanom, når begge kreftformer er knyttet til UV-bestråling? Og hvorfor får mennesker med medfødt kreftfremmende mutasjon i APC-genet oftere tykktarmskreft enn tynntarmskreft, når medfødte mutasjoner sitter i alle celler i kroppen? Jeg vil i denne artikkelen presentere to arbeider av Cristian Tomasetti og Bert Vogelstein m.fl., publisert i 2015 og 2017, og som diskuterer unøyaktigheter under kopiering av arvematerialet som en kilde til kreftfremkallende mutasjoner1,2. Til forskjell fra arv og miljø, som representerer «ytre» faktorer som disponerer for kreftutvikling, så er unøyaktigheter under kopiering av arvemateriale en iboende egenskap ved all DNA-replikasjon – og ikke bare årsak til sykdom men også selve kilden til livet som frembrakt ved evolusjon.
Vår søken etter å bekjempe kreft har en lang historie, se for eksempel ‘kjemoterapiens historie’ i Onkonytt 2016 nummer 2. Vår søken etter å forstå årsaken til kreft har en minst like lang historie, i håp om å finne effektiv behandling når årsak er kjent.
Elektronmikroskopisk bilde av stamceller (Åsmund Flobak)
Antall stamcelledelinger per vev per menneske betegnes lifetime stem cell divisions (lscd) og angis ut fra antall stamceller (s) og antall stamcellegenerasjoner (d) gjennom 80 år. Dersom det antas at en stamcelle deler seg og gir opphav til to datterceller, som igjen kan gi opphav til fire nye datterceller, som igjen kan gi opphav til åtte nye datterceller, så vil antall stamcelledelinger (og DNA-replikasjoner) under vevsdannende fase være 2+4+8+16+…+s, hvor s altså er antall observerte stamceller i det endelige organet. Summen av denne geometriske rekken av tall blir lik 2s-2, dvs det dobbelte antall stamceller i endelig vev minus 2. Etter at vevet er dannet går organet over i vevshomeostasefasen, og her vil antall stamcelledelinger være lik antall stamceller (s) multiplisert med antall stamcellegenerasjoner (d) gjennom hele livet, s x d. Totalt antall stamcelledelinger per vev blir således summen av antall stamcelledelinger i den første og andre fasen: lscd = 2s -2 + s x d = s(d+2)-2 (se figur 2).
Det er allment akseptert at arv og miljø kan disponere for kreft. Men ikke bare arv og miljø, også andre forhold er vist å disponere for kreft, som kroppshøyde. Hos kvinner er det vist at for kreft samlet er relativ risiko 1.16 for hver 10 cm ekstra høyde3, men med store variasjoner mellom kreftdiagnosene, og for eksempel langt mindre sammenheng mellom høyde og kreft ved kjent ytre faktor som røyking ved lungekreft. En logisk forklaring på denne observasjonen kan simpelthen være at flere celler betyr større totalrisiko for kreft, dersom det er slik at hver celle har en tilnærmet lik og iboende risiko for å bli en kreftcelle. (En annen historie er at denne sammenhengen mellom kroppsstørrelse og kreftrisiko ikke gjelder på tvers av arter4).
Bert Vogelstein og medarbeidere har i to artikler vist en sammenheng mellom antall stamcelledelinger som trengs for å lage et vev, og risikoen for kreft som utgår fra dette vevet. Ved hver stamcelledeling vil arvematerialet (DNA) kopieres, og ved hver slik kopiering er det en viss risiko for at små feil introduseres i DNA-sekvensen, som i så fall vil arves videre til alle kommende datterceller (anslagsvis cirka 3 mutasjoner per stamcelledeling). Vogelstein deler inn stamcelledelingen i to hovedfaser for hvert organ: 1) vevsdannelsen, hvor noen få celler i fosterlivet gir opphav til alle celler i det ferdige organet, og 2) vevshomeostasen, hvor stamceller kontinuerlig deler seg for å erstatte tapte celler. For større organer kan bidraget til antall stamcelledelinger være betydelig fra den første fasen, for eksempel lever sammenlignet med binyrer. For vev med høyere omsetning av celler vil bidraget til antall stamcelledelinger være betydelig fra den andre fasen, for eksempel tykktarmsepitel versus ørebruskceller. Vogelstein beregner antall stamcelledelinger som å bestå av summen av antall stamcelledelinger som trengs fra den første fasen (vevsdannelse) og den andre fasen (vevshomeostase), og beregner antall stamcelledelinger ut fra observert antall stamceller i det endelige vevet, sammenholdt med data på hvor ofte stamceller fornyes under vevshomeostasen i forskjellige vev.
Vogelstein har videre innhentet livstidsrisiko for kreft fra epidemiologiske databaser5,6. Figur 1 viser hvordan livstidsrisiko for 31 vevs- og subtyper av kreft korrelerer med antall stamcelledelinger lscd. Som det ses av figuren er det et relativt godt samsvar mellom de to variablene med Pearson korrelasjonskoeffisient 0,80, som er en god korrelasjon gitt alle forenklinger og mangelfulle data som ligger til grunn for analysen. Dette betyr at av de tre faktorene som bidrar til kreftmutasjoner (arv, miljø, tilfeldighet), så er sannsynligvis tilfeldige feil under DNA-kopiering en vesentlig del av bildet, faktisk vil en Pearson korrelasjonskoeffisient på 0,8 indikere at cirka 2/3 av alle kreftfremmende mutasjoner skyldes tilfeldighet. Det er imidlertid vesentlig å merke seg at dette ikke betyr at 2/3 av kreftforekomst skyldes tilfeldighet – for å utvikle kreft trengs flere kreftfremmende drivermutasjoner (engelsk: driver mutations) som sammen gir kreftsykdom. Det eksakte antall mutasjoner som trengs er ikke kjent, og varierer sannsynligvis fra kreftform til kreftform, og fra pasient til pasient, men er anslått til å være cirka 3 (lavere for flere hematologiske kreftformer). Tenkt eksempel: Hvis en pasient akkumulerer 2 kreftfremmende mutasjoner gjennom et langt liv på basis av feilkopiering av DNA og 1 kreftfremmende mutasjon fra et mutagen i miljøet så kan en kreftform som krever 3 kreftfremmende mutasjoner unngås ved å unngå miljøeksponeringen, siden de 2 mutasjonene som skyldes iboende egenskaper ved DNA-kopiering ikke alene ville være tilstrekkelig for å utvikle kreft.
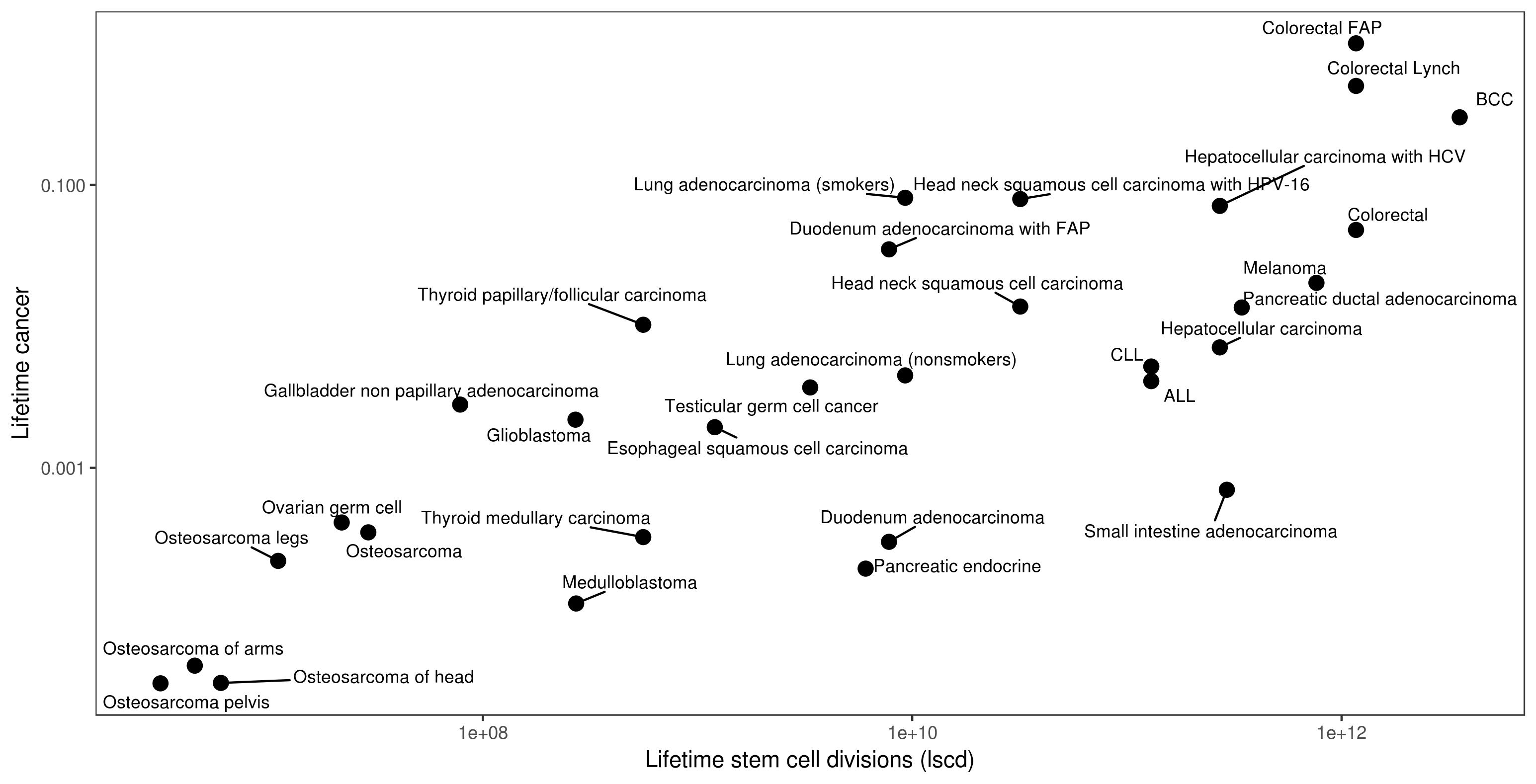
Figur 1 Livstidsrisiko for kreft mot antall stamcelledelinger i vevet.
Som det ses av figur 1 er det også slik at noen kreftformer i mindre grad kan “forklares” ut fra antall stamcelledelinger, og det er nærliggende å tenke at for disse kreftformene vil arv og/eller miljø være vesentlige deler av årsaken til kreftsykdommen. For eksempel ses det av figuren at for lungekreft så vil den gruppen pasienter som røyker ha en høyere forekomst av kreft enn den gruppen av pasienter som ikke røyker, noe som er lett å forstå siden tobakksrøyk er et mutagent stoff. Videre ses det at for kreftformer med kjent genetisk disposisjon for kreft, som for pasienter med tykktarmskreft og familiær adenomatøs polypose (mutasjon i APC-genet), så er kreftforekomsten mye høyere enn for pasienter uten en slik genetisk faktor.
Artikkelen fra 2015 ble blant annet kritisert for kun å se på en amerikansk populasjon, men samme korrelasjon 0,80 (95 % -intervall: 0,67-0,84) ble gjenfunnet når data for 69 land ble vurdert og publisert i 2017. I 2017-publikasjonen gikk forskerne videre og forsøkte ved hjelp av epidemiologiske data og sekvensering av tumor-DNA å kvantifisere bidragene fra arv versus miljø versus replikasjonsfeil. For de fleste kreftformer finner forskerne at feil under DNA-replikasjon er hovedårsak til kreftfremmende mutasjoner, med viktige unntak som lunge-, øsofagus-, hode-hals, føflekk- og ventrikkel-cancer.
Så hvilke konklusjoner kan trekkes fra funnene i disse to artiklene? Arbeidet kan blant annet bidra til å identifisere for hvilke kreftformer ytre faktorer (arv og/eller miljø) spiller en rolle, samt gi et tak for hvor stor effekt man kan forvente av for eksempel livstilsintervensjon. Forfatterne argumenterer også for at selv om primærforebygging av kreft er vesentlig for alle kreftformer med kjent ytre, ikke-arvelig faktor, så vil sekundærforebygging være vesentlig for både arvelig og “tilfeldighetsdrevet” kreft, hovedsakelig i form av tidlig diagnose etter oppstått kreftsykdom for å muliggjøre rask behandling.
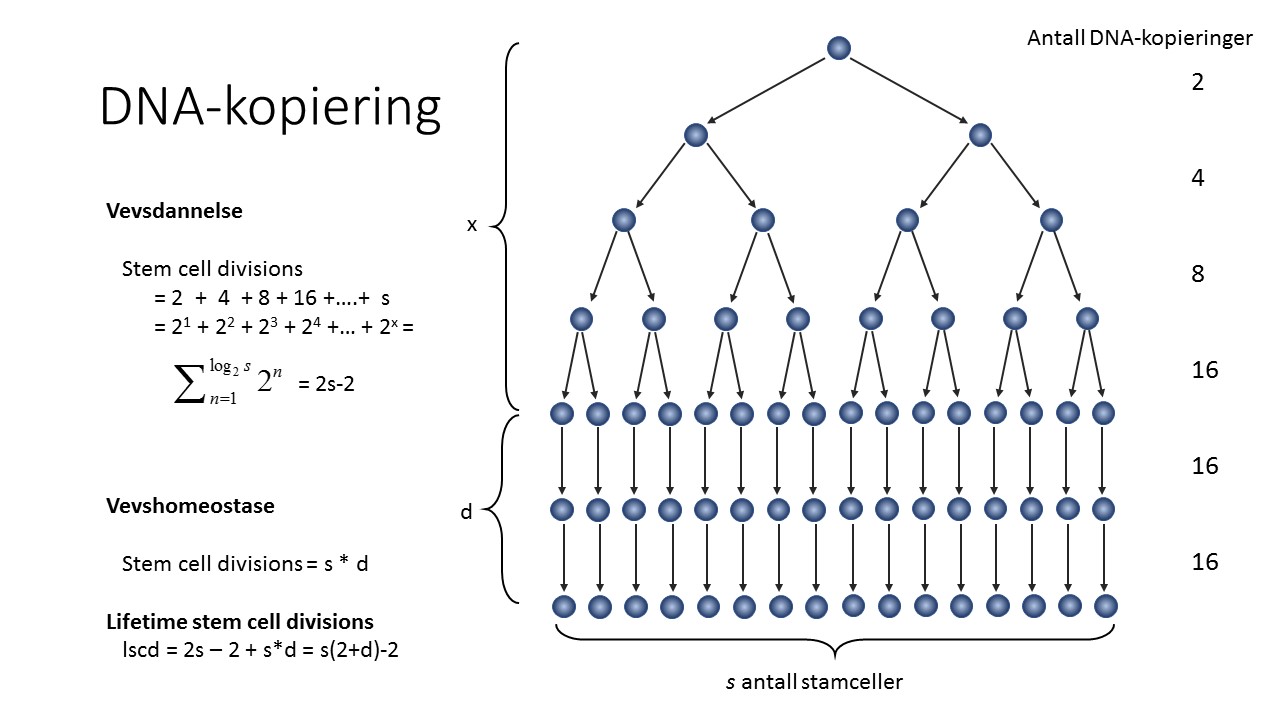
Figur 2: Antall DNA-kopier laget under vevsdannende og vevshomeostatisk fase. Hver figur i pilen representerer én DNA-replikasjon. Antall DNA-replikasjoner under vevsdannende fase kan enkelt beregnes hvis en kjenner antall stamceller i det endelige vevet (s). Antall DNA-replikasjoner under vevshomeostatisk fase vil være lik antall stamceller multiplisert med antall delinger per celle per livstid. Summen av antall stamcelledelinger fra vevsdannende og vevshomeostatisk fase vil være lscd = s(2+d)-2.
Pearson korrelasjonskoeffisient sier hvor stor andel av variasjonen i en variabel som kan “forklares” ut fra variasjonen i en annen variabel, og en verdi på 0 indikerer intet samsvar, mens en verdi på 1 tilsvarer perfekt samsvar, og en verdi på -1 tilsvarer perfekt samsvar men med negativt fortegn. En sammenheng i figuren som går fra nedre venstre til øvre høyre kvadrant gir positiv Pearson korrelasjon, mens dersom sammenhengen hadde gått fra øvre venstre til nedre høyre kvadrant ville Pearson korrelasjon vært negativ. Hva som er en god korrelasjon er forskjellig fra fagfelt til fagfelt, grovt sett vil det f.eks. i partikkelfysikkeksperimenter kunne være korrelasjoner tett mot 1 (eller -1), mens i biologiske forsøk kan verdier fra 0,5 anses akseptable. Kvadratet av korrelasjonskoeffisienten er et mål på hvor stor andel av variansen i y-aksen som forklares ut fra variansen langs x-aksen, dvs hvor sikkert kan man vite y-koordinaten for et punkt dersom man kjenner kun x-koordinaten (for to uavhengige variabler vil kjennskap til x-koordinaten ikke bidra til gjetning om y-koordinaten).
Referanser
- Tomasetti, C., & Vogelstein, B. (2015). Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science (New York, N.Y.), 347(6217), 78–81. doi:10.1126/science.1260825
- Tomasetti, C., Li, L., & Vogelstein, B. (2017). Stem cell divisions, somatic mutations, cancer etiology, and cancer prevention. Science (New York, N.Y.), 355(6331), 1330–1334. doi:10.1126/science.aaf9011
- Green, J., Cairns, B. J., Casabonne, D., Wright, F. L., Reeves, G., & Beral, V. (2011). Height and cancer incidence in the Million Women Study: prospective cohort, and meta-analysis of prospective studies of height and total cancer risk. The Lancet Oncology, 12(8), 785–794. doi:10.1016/S1470-2045(11)70154-1
- Abegglen, L. M., Caulin, A. F., Chan, A., Lee, K., Robinson, R., Campbell, M. S., … Schiffman, J. D. (2015). Potential Mechanisms for Cancer Resistance in Elephants and Comparative Cellular Response to DNA Damage in Humans. Jama, 84112(17), 1–11. doi:10.1001/jama.2015.13134
- National Cancer Institute, Surveillance, Epidemiology, and End Results Program; www.seer.cancer.gov.
- International Agency for Research on Cancer (IARC) (http://ci5.iarc.fr/CI5-X/Pages/download.aspx)