
Hildegunn Høberg-Vetti, leder, Regionalt kompetansesenter for arvelig kreft, Haukeland universitetssjukehus, Helse Vest
Lars Fredrik Engebretsen, seksjonsoverlege, Senter for medisinsk genetikk og molekylærmedisin, Haukeland universitetssjukehus


Hildegunn Høberg-Vetti
Innledning
Arvelig kreft kan defineres som kreftsykdom som er forårsaket av en nedarvet genfeil i et kreftgen med høy penetrans, dvs at de fleste med genfeilen utvikler sykdom(1). Vanligvis er arvegangen autosomal dominant, men autosomal recessiv arvegang kan også forekomme. Begrepet familiær kreft bruker vi i dag om familier med økt kreftopphopning, men uten påvisbar genfeil i et høypenetrant kreftgen. Det antas at slike tilfeller oftest dreier seg om multifaktoriell arv, der både flere lavpenetrante genvarianter og miljøfaktorer er av betydning for kreftuviklingen(2).
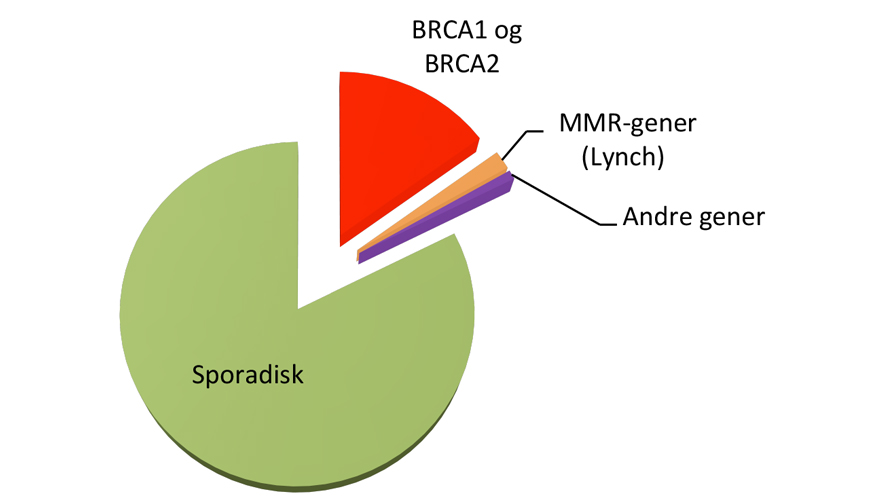
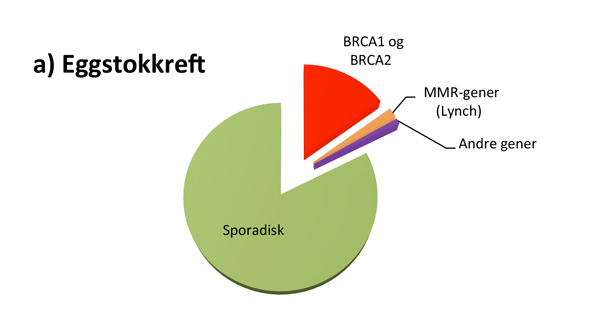
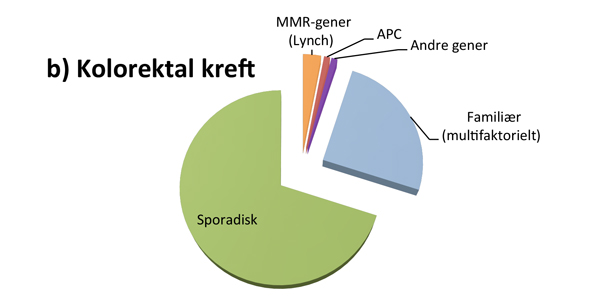
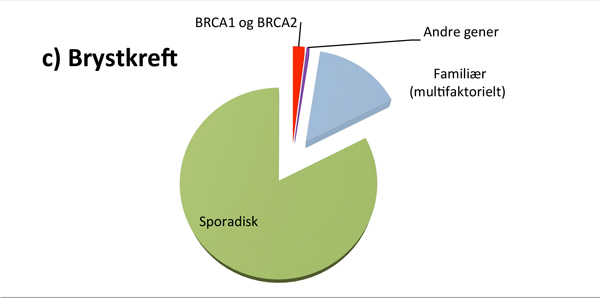
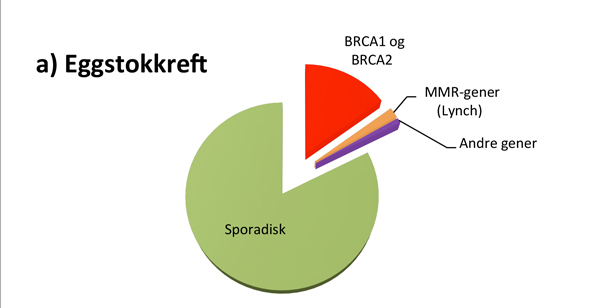
Hvor stor andel av kreftpasientene som har den monogent arvelige typen, varierer fra kreftform til kreftform (se figur 1). For eksempel er det rapportert at opptil 25% av alle feokromocytomer og drøye 20% av alle tilfeller av epitelial eggstokkreft kan skyldes en medfødt genfeil, mens 2-3% av alle med brystkreft og 3-5% av alle med tarmkreft har en tilgrunnliggende høypenetrant genfeil(3-5).
Grunnlaget for diagnostisk gentesting av kreftpasienter er todelt: Det ene er at man kan identifisere familier med arvelig kreft og dermed identifisere flere personer med høy risiko for kreft. Ved å tilby slike høyrisikopersoner intensivert kontrollopplegg og risikoreduserende kirurgi, har man en unik mulighet til å forebygge kreft og kreftrelatert død(6-10). Det andre aspektet er at gentestsvaret kan ha betydning for prognose og behandling av den aktuelle pasientens kreftsykdom(11). Indikasjonsstillingen i det enkelte tilfelle må sees i lys av dette.
Gentester
Den raske teknologiske utviklingen og identifisering av nye kreftgener har ført til at repertoaret av diagnostiske gentester stadig øker(12). Med persontilpasset medisin vil gentesting etter hvert bli en del av alle onkologers kliniske hverdag, og det er viktig at man kjenner til mulighetene, men også begrensningene ved de mest vanlige gentestene for arvelig kreft. Disse vil derfor bli nærmere omtalt i det følgende.
Mismatch Repair genene: MLH1, MSH2, MSH6 og PMS2
Den hyppigste formen for arvelig tarmkreft er Lynch syndrom, forårsaket av en nedarvet genfeil i et DNA-reparasjonsgen av gruppen mismatch repair (MMR)-gener; MLH1, MSH2, MSH6 og PMS2. Det er anslått at ca. 3% av alle med kolorektal kreft har Lynch syndrom. Genfeil i MMR-genene er også forbundet med høy risiko for endometriekreft hos kvinner og noe økt risiko for flere andre kreftformer (bl.a. kreft i magesekk, tynntarm, pankreas, urinveier og eggstokker)(7).
For å identifisere flere pasienter med Lynch syndrom bør svulstvevet analyseres for mikrosatellittinstabilitet (MSI) og immunhistokjemi (IH) av MMR-gener hos alle personer med tarmkreft før 50/60 års alder, pasienter med flere Lynch assosierte kreftsykdommer og pasienter med tarmkreft og positiv familiehistorie(13). De fleste svulster ved Lynch syndrom vil vise høy grad av mikrosatellittinstabilitet («MSI-High»), og IH vil kunne gi en indikasjon på hvilket MMR-gen som er involvert. (NB! Også 15 % av sporadiske tarmkreftsvulster kan vise høy grad av mikrosatellittinstabilitet, vanligvis forårsaket av somatisk hyper-metylering av MLH1 promotor og/eller BRAF-mutasjon.) Dersom resultatet av MSI- og/eller IH-analysen indikerer at pasientens tarmkreft kan være ledd i Lynch syndrom, bør pasienten henvises videre til medisinsk genetisk avdeling for genetisk veiledning og diagnostisk gentest av MMR-genene i blodprøve.
APC og MUTYH
Klassisk familiær adenomatøs polypose (FAP) skyldes genfeil i APC-genet, men utgjør mindre enn 1% av alle med kolorektal kreft(5). I opptil 20% av tilfellene er genfeilen nyoppstått hos pasienten, og familiehistorien vil da være negativ. Polypputviklingen starter vanligvis i tenårene, og ubehandlet er risikoen for kolorektal kreft tilnærmet 100%. I tillegg til APC-genet finnes det flere andre, mer sjeldne gener, forbundet med polyposetilstander i tarmen, bl.a. MUTYH-genet som gir en autosomal recessiv tarmpolypose(5).
BRCA1 og BRCA2
I Norge har opptil 20-25% av alle kvinner med eggstokkreft og 2-3% av alle kvinner med brystkreft genfeil i BRCA1 eller BRCA2 genet, med høyest prevalens på Sør-Vestlandet(4). Den relativt høye prevalensen av genfeil i Norge skyldes såkalte foundermutasjoner; dvs mutasjoner som har oppstått for utallige generasjoner siden, og som har fått en viss utbredelse i befolkningen. Pga. dette har kriteriene for BRCA-gentesting vært mer liberale i Norge sammenlignet med de fleste andre land. For gjeldende kriterier henvises det til Nasjonalt Handlingsprogram for brystkreft(14).
Hvis en ikke tar hensyn til familiehistorien, er risikoen for å utvikle brystkreft innen 70 års alder hos en kvinne med BRCA1– genfeil ca. 60%, mens risikoen for kontralateral brystkreft er over 80% (15-17). Brystkreften er ofte høygradig malign, av trippel negativ type. Tilsvarende risiko for eggstokkreft er ca. 30-60% (15, 16). Ved genfeil i BRCA2-genet er risikoen noe lavere; ca. 30-55% for brystkreft, ca. 5-15% for eggstokkreft og ca. 60% for kontralateral brystkreft(15, 16). Imidlertid er det også økt risiko for flere andre kreftformer hos personer med BRCA2-genfeil, som pankreaskreft, prostatakreft (ofte relativt aggressiv type) og brystkreft hos menn(18).
Kvinner med BRCA-genfeil kan være aktuelle kandidater for målrettet terapi med PARP-hemmere. Den første PARP-hemmeren ble godkjent av FDA i 2014 for pasienter med eggstokkreft(19). I Norge pågår det kliniske forsøk med PARP-hemmere både for pasienter med brystkreft og pasienter med eggstokkreft(20, 21).
CDKN2A og CDK4
Ved familiær opphopning av malignt melanom er det aktuelt å undersøke CDKN2A-genet og CDK4-genet(22). Genfeil i ett av disse genene gir en betydelig forhøyet risiko for utvikling av malignt melanom, og kan også være forbundet med dysplastisk nævus syndrom. Pasientens hudtype og solvaner vil også spille inn på den enkeltes risiko for å få melanom(23, 24). Det er også rapportert høyere risiko for pankreas-kreft, lungekreft og andre tobakksrelaterte kreftformer hos personer med CDKN2A-genfeil(25).
Lavpenetrante gener forklarer imidlertid det meste av den familiære opphopningen også ved malignt melanom, særlig gener som regulerer hudtype og hårfarge – f.eks. MC1R-genet(5). Kun en mindre andel (< 10%) av familiene med økt forekomst av melanom har en påvisbar høypenetrant genfeil.
MEN1 og RET
Arvelig sårbarhet for svulster i endokrine organ kan skyldes genfeil i mange ulike gener.
Genfeil i MEN1-genet gir multiple endokrine neoplasier type 1 (MEN1). Ved MEN1-tilstanden kan det bli svulster i en rekke endokrine organ. Svulstene er ofte godartede, men de produserer hormoner. De hyppigst affiserte endokrine organer er parathyroidea (gir hyperparathyroidisme med hyperkalsemi), hypofysen og pankreas. I pankreas kan svulstene bli ondartede. Genfeil i MEN1-genet har høy penetrans, rundt 95% ved 40 års alder(5, 26).
Genfeil i RET-onkogenet gir syndromet multiple endokrine neoplasier type 2 (MEN2A/MEN2B). Vanligste funn ved MEN2-tilstanden er medullært thyroideakarsinom (MTC) som kan debutere i svært ung alder. Det er derfor aktuelt med forebyggende thyroidektomi i barnealderen, og indikasjon for prediktiv gentest av barn allerede i spedbarnsalder. Genfeil i RET har høy penetrans for MTC, noe avhengig av genfeilen (genotype-fenotype korrelasjon)(5).
{kind=link}
Sjeldne kreftsyndromer
Det finnes en rekke kjente arvelige kreftsyndromer, eller tumorsyndromer, som gjerne gir et helt spesifikt tumorspektrum i de affiserte familiene. Noen av syndromene diagnostiseres pga spesifikke kliniske trekk hos den enkelte pasient. Et eksempel er PTEN Hamartoma tumor syndrom (Cowden syndrom). Genfeil i PTEN-genet gir i tillegg til økt risiko for maligne og benigne svulster i bryst, skjoldbruskkjertel og livmor også en rekke andre fenotypiske trekk som kan være tilstede i mer eller mindre grad; makrocephali, papillomatøse lesjoner i hud og slimhinner, lærevansker mm(5). Andre kreftsyndromer kommer til genetisk utredning pga. spesielle kombinasjoner av ulike kreftsykdommer i familien. Li-Fraumeni syndrom, forårsaket av genfeil i TP53-genet er et eksempel på dette. Personer med medfødt genfeil i TP53-genet har svært høy risiko for kreft i ung alder, spesielt sarkomer, premenopausal brystkreft, hjernesvulst, adrenokortikalt karsinom og akutt leukemi(5).
I mange familier er det opphopning av ulike kreftsykdommer uten at man kan gjenkjenne noen av de beskrevne kreftsyndromene, og gentesting er ofte uten resultat. I noen av disse familiene kan kreftopphopningen skyldes ren tilfeldighet (jf. at kreft er en vanlig sykdom), mens andre familier trolig har en økt sårbarhet for kreft pga. en kombinasjon av lavpenetrante genvarianter og miljømessige forhold, jf. ovenfor. Men en liten andel av de hardest belastede familiene kan også tenkes å ha genfeil i et svært sjeldent kreftgen, der man ennå ikke har kartlagt årsakssammenhengen.
For de fleste kreftformer må man regne med at det finnes en undergruppe som er arvelig. Fokuset frem til nå har vært på de store kreftgruppene, og gentesting for arvelig brystkreft, tykktarmskreft og føflekkreft er blitt tilgjengelig i klinisk praksis. Like fullt er det viktig å være oppmerksom på familier med opphopning av andre kreftformer. Nyrekreft, hjernesvulst og pankreaskreft er eksempler på sykdommer som forekommer familiært, med tilsynelatende autosomal dominant arvegang, men hvor årsaksgenene foreløpig er dårlig kartlagt. Ny sekvenseringsteknologi («dypsekvsensering») gir større muligheter for å påvise den genetiske årsaken i disse sjeldne familiene – forutsatt at det er tilgjengelig DNA fra en eller flere av de syke. Kreftpasienter som tilhører slike tungt belastede familier bør tilbys henvisning til genetisk veiledning og utredning. Det kan da være aktuelt å benytte seg av paneltester (se under), evt å gjøre utvidet søk etter ukjente gener ved hjelp av eksom/helgenomsekvensering.
Multigen paneltester
Dypsekvenseringsteknologien har gitt bedre muligheter for å undersøke mange gener parallelt i en prøve. Slike paneltester er nå i planleggings- og etableringsfasen ved flere medisinsk-genetiske laboratorier i Norge. F.eks. er det de senere år identifisert flere gener som kan gi tarmpolypose, men der kun en svært liten andel av pasientene har genfeil i hvert enkelt gen(27, 28). Ved å undersøke alle disse genene i én analyse har man større mulighet for å finne den tilgrunnliggende genfeilen hos flere pasienter.
Men selv om teknologien gir nærmest ubegrensede muligheter for hva man kan undersøke, må det alltid veies opp mot hvilken nytte man har av resultatet. Paneltester som omfatter høypenetrante kreftgener vil ha en helt annen klinisk nytteverdi enn paneltester som inkluderer moderat og lavpenetrante kreftgener. Sistnevnte benyttes i dag i forskningssammenheng, men har foreløpig ingen plass i den klinisk diagnostiske virksomheten.
Genetisk utredning og
persontilpasset medisin
I en tid som er preget av en rivende teknologisk utvikling, er det viktig å minne om at det er tre hovedelementer i enhver genetisk utredning:
- Familieanamnese
- Pasientens egen sykehistorie og
kliniske funn - Genetiske laboratorieundersøkelser.
De to første punktene inngår i god klinisk praksis og danner grunnlaget for å vurdere om det er indikasjon for gentesting av den enkelte pasient. Innføringen av persontilpasset medisin i helsevesenet vil sannsynligvis føre til økt bruk av genetisk diagnostikk i kreftutredning og -behandling(29). Økningen vil først og fremst gjelde kartlegging av genetiske endringer i selve svulsten, men også nedarvede genetiske varianter vil bli identifisert i større grad enn tidligere. Familiehistorien og pasientens klinikk vil være avgjørende for tolkning av disse variantene, og helt nødvendig for å sikre at den nye teknologien kommer best mulig til nytte for kreftpasientene.
Referanser:
1. Rahman N. Realizing the promise of cancer predisposition genes. Nature 2014; 505: 302-8.
2. Foulkes WD. Inherited susceptibility to common cancers. The New England journal of medicine 2008; 359: 2143-53.
3. Neumann HP, Bausch B, McWhinney SR, et al. Germ-line mutations in nonsyndromic pheochromocytoma. The New England journal of medicine 2002; 346: 1459-66.
4. Moller P, Hagen AI, Apold J, et al. Genetic epidemiology of BRCA mutations–family history detects less than 50% of the mutation carriers. European journal of cancer 2007; 43: 1713-7.
5. Hodgson S; Foulkes WD; Eng C; Maher ER. A Practical Guide to Human Cancer Genetics. Fourth Edition. London: Springer-Verlag; 2014.
6. Jarvinen HJ, Mecklin JP, Sistonen P. Screening reduces colorectal cancer rate in families with hereditary nonpolyposis colorectal cancer. Gastroenterology 1995; 108: 1405-11.
7. Vasen HF, Blanco I, Aktan-Collan K, et al. Revised guidelines for the clinical management of Lynch syndrome (HNPCC): recommendations by a group of European experts. Gut 2013;
62: 812-23.
8. Evans DG, Kesavan N, Lim Y, et al. MRI breast screening in high-risk women: cancer detection and survival analysis. Breast cancer research and treatment 2014; 145: 663-72.
9. Rebbeck TR, Friebel T, Lynch HT, et al. Bilateral prophylactic mastectomy reduces breast cancer risk in BRCA1 and BRCA2 mutation carriers: the PROSE Study Group. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2004; 22: 1055-62.
10. Ingham SL, Sperrin M, Baildam A, et al. Risk-reducing surgery increases survival in BRCA1/2 mutation carriers unaffected at time of family referral. Breast cancer research and treatment 2013; 142: 611-8.
11. Lee JM, Ledermann JA, Kohn EC. PARP Inhibitors for BRCA1/2 mutation-associated and BRCA-like malignancies. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO 2014; 25: 32-40.
12. Norsk portal for medisinsk-genetiske analyser. www.genetikkportalen.no (31.05.2015)
13. Nasjonalt handlingsprogram med retningslinjer for diagnostikk, behandling og oppfølging av kreft i tykktarm og endetarm. Oslo: Helsedirektoratet, 2015.
14. Nasjonalt handlingsprogram med retningslinjer for diagnostikk, behandling og oppfølging av pasienter med brystkreft. Oslo: Helsedirektoratet, 2015.
15. Mavaddat N, Peock S, Frost D, et al. Cancer risks for BRCA1 and BRCA2 mutation carriers: results from prospective analysis of EMBRACE. Journal of the National Cancer Institute 2013; 105: 812-22.
16. Brohet RM, Velthuizen ME, Hogervorst FB, et al. Breast and ovarian cancer risks in a large series of clinically ascertained families with a high proportion of BRCA1 and BRCA2 Dutch founder mutations. Journal of medical genetics 2014; 51: 98-107.
17. Moller P, Maehle L, Vabo A, et al. Age-specific incidence rates for breast cancer in carriers of BRCA1 mutations from Norway. Clinical genetics 2013; 83: 88-91.
18. Moran A, O’Hara C, Khan S, et al. Risk of cancer other than breast or ovarian in individuals with BRCA1 and BRCA2 mutations. Familial cancer 2012; 11: 235-42.
19. Olaparib approved for advanced ovarian cancer. Cancer discovery 2015; 5: 218.
20. REK-Vest ref.no 2014/924
21. REK-Vest ref.no 2013/1487
22. Nasjonalt handlingsprogram med retningslinjer for diagnostikk, behandling og oppfølging av maligne melanomer. Oslo: Helsedirektoratet, 2015.
23. Cust AE, Harland M, Makalic E, et al. Melanoma risk for CDKN2A mutation carriers who are relatives of population-based case carriers in Australia and the UK. Journal of medical genetics 2011; 48: 266-72.
24. Puntervoll HE, Yang XR, Vetti HH, et al. Melanoma prone families with CDK4 germline mutation: phenotypic profile and associations with MC1R variants. Journal of medical genetics 2013; 50: 264-70.
25. Helgadottir H, Hoiom V, Jonsson G, et al. High risk of tobacco-related cancers in CDKN2A mutation-positive melanoma families. Journal of medical genetics 2014; 51: 545-52.
26. Thakker RV, Newey PJ, Walls GV, et al. Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1). The Journal of clinical endocrinology and metabolism 2012; 97: 2990-3011.
27. Palles C, Cazier JB, Howarth KM, et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nature genetics 2013; 45: 136-44.
28. Weren RD, Ligtenberg MJ, Kets CM, et al. A germline homozygous mutation in the base-excision repair gene NTHL1 causes adenomatous polyposis and colorectal cancer. Nature genetics 2015; 47: 668-71.
29. Persontilpasset medisin i helsetjenesten. Rapport fra nasjonal utredning 2013/2014. http://www.helse-sorost.no/fagfolk_/forskning_/persontilpasset-medisin_/Documents/rapport_persontilpasset%20medisin.pdf (31.05.2015)